大腸菌やプラスミドDNAについて割と細かい点を見ていましたが、「めちゃ甘タンパク質・ソーマチンを作ってみよう」講座の本筋に戻って進めていきましょう。
ちなみに前回のOriの違いやら菌株の違いやらは、入門編を逸脱しているばかりか、たまに生命系専攻の学生ですらおざなりにして適当に使ってることすらある話なので、バッチリ理解されたら正直生命系の大学生よりちゃんとこの辺の話を知っているといえるぐらいのレベルになるかと思います。
逆にいうと、ややこしくはあるけれど、やっぱり数学・物理などとは違い、整理さえできれば誰でも理解できる話になっているともいえるかもしれない感じですね。
(大学レベルの数学や物理学は、高校範囲の知識から積み上げていかないと、いきなり説明されても絶っ対に理解できないけど、生命科学系はやっぱり、知識の積み立てが全くないとはいわないけれども、良くいえば圧倒的に理解しやすく、悪くいえば理論の構築&展開というより単なる現象論の描写に過ぎないので、厳密性に欠ける浅い話が多いという感じ。)
まぁそんな御託はともかく、今回はプラスミドを大腸菌に入れて、増やして、回収という作業について見てみるとしましょう。
ちょうど毎度大変丁寧なコメントをいただけるアンさんから、「制限酵素とライゲーション…分かりやすい図入りの記事があったはずだが、どこだったろうか……参考リンクがあれば助かる」的なメッセージをいただいていました。
確かにどの記事で見ていたかのリンクがあった方が親切ですね!
まぁ何記事かにわたって見ていたこともあるので「ズバリこれ」とはいかないこともありますが、一番メイン部分を説明していた記事を、参照先として今後のフローチャートにはリンクも貼るようにしましょう。
ナイスアドバイス、どうもありがとうございました…!
【大腸菌にタンパク質を作ってもらおう!】
1. 遺伝子DNAをゲットする!⇒済み!(遺伝子を注文しよう!)
2. そのDNAを、制限酵素とDNAリガーゼを使って、プラスミドに導入する(クローニング)!⇒済み!(図を見れば一発?!改めて、分かりやすくDNA切り貼りの仕組みを紹介!)
3. その遺伝子組込みプラスミドDNAを大腸菌にぶち込む(形質転換)!⇒済み!(たった数日で1000万円分のモノが作れる、楽しい作業)
4. DNAがぶち込まれた大腸菌の選別!⇒済み!(遺伝子をぶち込まれたやつだけが生き残れる、サバイバルゲーム!)
5. 選ばれた「DNAがぶち込まれた大腸菌」をひたすら増やそう!←今ココ
6. タンパク質合成のスイッチON!
7. 満を持して、目的タンパク質の収穫!
8. さすがにそのまんまでは大腸菌まみれで汚いので、キレイに精製しよう!
→見事、手元には大量の純品タンパク質が!やったね!!
直近3回ぐらいの記事で、用途に応じてそれぞれDNAクローニング用のプラスミド・大腸菌株と、さらにタンパク質合成用のプラスミド・大腸菌株などがあってどうたらとかを見ていたわけですが、結局、今回の一連の実験ではそんなに大量のDNAが必要とならないため、DNAクローニング用プラスミドの出番はなく、大腸菌内でのコピー数が少ないので回収できるDNA量も少ないpET-15bというタンパク質合成用に特化したプラスミドDNAを使うだけで十分という話でした。
しかし、流石にライゲーション産物を直でタンパク質合成用大腸菌(B株)にぶち込むことは乱暴すぎるので普通はせず(もちろん、最終的にはそれが目的なわけですけどね)、最初はDNAクローニング用の大腸菌(K株)にぶち込んで、菌を増やして純品プラスミドを回収&シークエンスの確認をし、その後、その「配列が完璧に確認でき、量も十分手元にあるプラスミドDNA」を改めて最終目的のB株にぶち込むのが賢明なやり方といえましょう。
(直接B株にいくのが良くない理由は、ライゲーション産物には、もしかしたら変な形でつながってしまったものや、制限酵素で切れずに空ベクターのまま存在するものが紛れているかもしれないからですね。
そんな、何が入ったか分からないまま実験を進めるのは、あまりにも危険というか見切り発車がすぎる、というわけです。)
ということで、上記手順の3~5を済ませたら、手元には一旦プラスミドDNAのクローンが得られるわけですけど、シークエンスを読み、1塩基のズレもなく正しい配列が入ったことを確認したら、改めてまた3番の形質転換に戻り、今度は純品プラスミドを最終目的のB株に入れる、って形になるわけですね。
つまり、上記まとめは、実はかなり端折ったフローチャートだったという形です(挙げていない点では、4と5の間で、グリセロールストックを作ることなんかもありますね)。
…と、改めて前半ステップのおさらいをしましたが、ようやくステップ5の、DNAを増やす話についてですね。
簡単にまとめると、大腸菌を栄養たっぷりの液体培地で増やして、プラスミドは大腸菌が分裂するときに勝手に、自動的に同じように増えてくれますから、十分大腸菌が増殖したところで「ありがとなっ!もう用はないから死んでいいよ」と、大腸菌をぶっ壊してプラスミドDNAだけをゲットするという、そんな実験です。
この、大腸菌からプラスミドを抽出する作業をミニプレップ(略称ミニプレ…もちろん日本限定の略称ですが)と呼んでおり、これはプラスミドDNA・大腸菌の生育・分かりやすい試薬を使った処理による菌体からのDNAの精製・(最近の改良版では使わないやり方も多いものの)フェノクロ・エタ沈(この記事とかでも触れていた、分子生物学で一番基本的な実験)などなど、生命科学系研究で必要となる基本知識・手順が完全に網羅された、初学者用にうってつけのチュートリアル実験みたいな存在で、学生用の実験講座なんかで最高の題材といえる感じになっています。
簡単に検索しても、学生用の講義資料みたいなのが見つかりました。
www.cc.kochi-u.ac.jp
こちら(↑)の高知大学のページ、僕が学生の頃から存在している、まさに僕も学生の頃参考に眺めた記憶のある記事ですが、もう20年前の記事とか、今の学生の立場からしたら、僕が学生だった頃で置き換えて考えてみると、当時の僕が1980年代の記事を見るのと同じ時間間隔(感覚)になってるって、それマジ…?!
何か時間経過がバグってる気がしますが、まぁやる手順なんかは概ねこの記事にある通りですね(一部細かい点は違うけど、大体同じ)。
ただ、これは学生向けの、安価で行えるけど多少手間がかかる&クオリティが低いプラスミド回収法で、実際の研究室レベルでは、多くの場合が、Qiagen(我々日本人はキアゲンと呼んでますが、英語だとカイアジェン)という、特にこの類のDNAやRNA抽出試薬で最大手の会社が出しているminiprep kitを使っていることが多いと思います。
(日本語版の実験手順(プロトコール)のPDFファイルがこちら↓
https://www.qiagen.com/de/resources/download.aspx?id=8b192d12-0775-4af4-8416-27897981cc81&lang=ja-JP)
まぁいちいちPDFを開いて見るのも面倒なので、最早何万回もミニプレをしたこの僕がめっちゃ簡単にまとめると…
- (上記全体フローチャートのステップ4で)プレートに生えてきた大腸菌を、爪楊枝でピックアップ
- 大体2 mLぐらいの液体培地(アンピシリン入り)を入れた試験管にピックした菌を移し(ただ爪楊枝を液体にチョンとつけるだけ)、一晩、37℃で、立てたチューブをガシャガシャ揺らせる培養器で飼う
(培養器は、大小さまざまなサイズがありますが、小型のだとこういうのですね↓

https://taitec.net/product/br-23um・mr/より - 翌日、透明だった培地は菌の増殖で完全に濁っているので、例の親指サイズのチューブに培養液を移し、高速で回せる遠心機にかける
(遠心機は、これも各社から色々なのが出ていますが、こういう、チューブを24本並べて回せるのがスタンダードでしょう↓
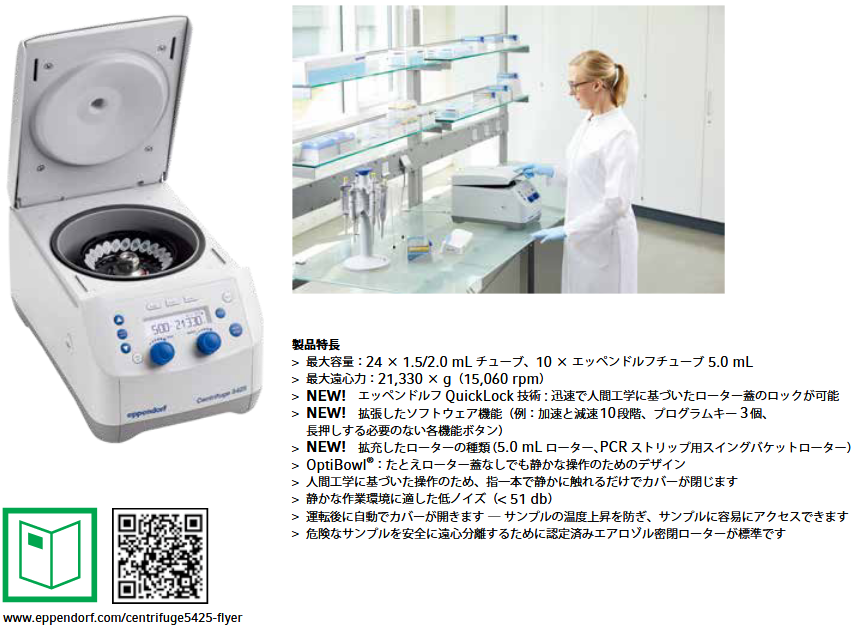
https://www.eppendorf.com/fileadmin/Countries/Japan/files/2020_MicroCentrifuge.pdfより - 大腸菌は結構重いので、遠心力でチューブの底に沈む。欲しいのはプラスミドを抱え込んでいる菌なので、不要な遠心上清(培地)は捨てる
- 菌体を、250 μL(=0.25 mL)の試薬P1(説明が面倒ですが、これ系の液体試薬を、業界人は「バッファー」と呼んでいます。pHが一定に保たれる性質があるからバッファー(緩衝液)と呼ぶわけですが、特に緩衝能がなくても、正直液体の試薬なら何でもかんでもバッファーって呼ぶことが多いですね)で懸濁する
- しっかり懸濁できたら、同じく250 μLのBuffer P2を加える
- チューブのフタを閉じ、何回か上下ひっくり返して混ぜる
(→大腸菌が、強アルカリ性のP2のおかげで完全に溶けて、めっちゃネバネバになる!ま、いうなれば、ちょうど鼻水みたいな感じですね) - しっかり溶けるまで2-3分待つことが多いけど、別に待たなくてもそんなに回収量は変わらない印象。
よくいわれるポイントとして、ここで待ちすぎると、大腸菌の染色体DNAもズタズタに分解されて溶け込んできてしまうので、長すぎるアルカリ処理は厳禁 - 上述の通りずっと溶かし続けても良くないので、酸性溶液であるBuffer N3を350 μL加えて、中和する。
ここだけなぜか350 μLなので、ピペットの設定を変え忘れないように注意(分かりやすく、加えるバッファーの量は250→250→250になるように変更&調節してくれればいいのに…)
(→N3を加えたら、鼻水状態だった溶液が、一気に真っ白なおからみたいな成分を含むさらさらな溶液に変貌を遂げる!変わらなかったら、何か間違っているということなので、実験は失敗) - これで、菌がぶっ壊れてプラスミドが大量に溶液内に放出されてきたことになるが、大腸菌の残りカスである白いおからが邪魔なので、またまた遠心して漂ってるカスを底に沈める
- 最高スピード(大体13,000 rpm=1分で1万3000回転=1秒で200回転以上!)で10分間遠心したら、今度はステップ4とは違い、必要なのは上清の液体であることに注意!(沈殿は細胞のカス。英語ではcell debris)
- 液体部分を、DNAを結合する力のある特別なカラムと呼ばれるチューブに通す。
(カラムは、こんなやつ(↓)。なぜかAmazonにも売ってました(笑)

https://www.amazon.com/Universal-Columns-Isolation-Extraction-Purification/dp/B07YLF7JR8より - Buffer PEというWashバッファーで2回ほどカラムを洗って、DNA以外の余計な成分を流し捨てる(カラムにPEを加えて、1分間遠心するだけ)。
- Buffer EBという溶出バッファーを加えれば、カラムに結合していたDNAが流れ落ちてくるので、これをキレイな新品チューブで回収して、無事、純品DNAをゲットだぜ!
…という流れで、まぁ細々した説明で長くなりましたが、普通に菌をスピンして、P1→P2→N3→スピン→カラム→ウォッシュ→エリューション(Elution;溶出)という、極めて単純なる流れです。
何となくのイメージでも明らかかもしれませんが、この実験ではカラムが一番特殊な物質で、これが結構高価なので、高知大学のプロトコールだと、学生実験ごときにカラムは使わせられねぇ、って感じで、P1P2N3(名前も入れる容量も、Qiagenとは違うみたいですが)処理して遠心した後は、フェノクロ→エタ沈するようになっていますね。
(記事では「フェノクロは省略することも多い」と書かれていますが、経験上、フェノクロをしないとクッソ汚い低品質のDNAになるので、これは絶対すべきだと思います。
…まぁ、学生実験ならクオリティは気にしないので省略しても問題ないかもしれませんが、フェノクロもしないミニプレDNAだと、シークエンシング業者に送っても、多分「DNAの品質が低すぎて読めませんでした」という結果が返ってきます。)
一方、カラムを使えば、めっちゃくちゃキレイなDNAが得られるので、簡単かつ便利で品質も高いと、ちょっと値が張る以外はいいことばかりです。
pUC系のクローニングベクターを入れた大腸菌の場合、ミニプレで大体20 μgぐらいのプラスミドDNAが回収できますかね。
pET系プラスミドだとまぁそれより大分少なくなりますが、それでもマイクログラムレベルのDNAは余裕で得られます。
まぁマイクログラムとかいわれても何も分からんて、って話かもしれませんが、この記事で見ていたように、シークエンス業者に送る必要があるのは500 ng=0.5マイクログラムとかそのぐらいなので、シークエンスを読んでも、まだまだ十分な量のDNAが残っており、これを使って続いてはB株の形質転換を行う、って話になるわけですね。
なお、Qiagenのキットはめっちゃ高いので、バッファー類は完全に自作し、カラムも、ちょうど上の写真でもAmazonのページを貼っていましたが、各社がカラム単独をより安価で販売してくれているので(僕が買ってるのはさっきの画像の会社ではないですけど)、別個で買っています。
この優れたカラム法ミニプレップを開発したのはQiagenなのに、やり方だけパクってQiagenには1円も払っていないのは何とも不届き者ですが、まぁ多くの研究室で自作バッファー&サードパーティーのカラムを使う方法で節約している感じですね。
実際、Buffer P1, P2, ..., EBまでQiagenのキットで使われているバッファー組成は、有志の手により全て明らかになって公開されています。
openwetware.org
もう特許とかは切れてるのかな?
Qiagenにしたら苦労して開発した試薬の成分を勝手に公開されるとかたまったものじゃないかもしれませんが、まぁリッチな研究室は普通に今でもQiagenキットを買うこともありますし(僕も、ミニプレキットはまぁ買いませんが、他の、Qiagenにしかない特殊な実験キットなんかは、たまにお世話になります)、勝手にプロトコールを使ってもしっかり感謝の念を送ればOKでしょう(適当)。
…って、よく見たら上のページにはなぜかEBの組成が載っていませんでしたが、「Qiagen Buffer EB」で検索したら、何気に、Qiagen社が自ら組成を公開してましたね(笑)。
www.qiagen.com
やはり恐らくもう特許は切れていて、自ら公開している感じなんでしょうか。
まぁ、試薬会社は結構ボッているので、売り上げとか利益とかも凄いようですし(Thermo Fisherの成長とかヤバイですしね)、人類の発展にも役立つ公共性のある財産ですから、特許が切れたものはガンガンシェアしてみんなで共有できるのが理想といえるかもしれませんね。
ということで、DNAの増やし方というか回収法・ミニプレについて見てみましたが、もうちょい補足で似た話をちょっとだけ触れようかなと思っています。
多分、少しだけ続く…。