前回は、「よく分かる解説!」と大見得を切ったくせに、やや丁寧さに欠ける、微妙な説明になってしまっていました。
正直、書いてても「いや『とても分かりやすく』はないわな」と自覚していたのですが、実はちょっと時間がなくて、昨日時点では改善を諦めていた感じでした。
しかし、今回はマジでめちゃくちゃ分かりやすいです。
ということで、改めて仕切り直しで、ほぼ1から説明を再開させていただくとしましょう。
(じゃあ前回の記事は何だったんだよ(笑)、って話になるかもしれませんが、一応、ジデオキシヌクレオチドとかその辺の基礎知識には触れられたので、その辺はもう前提に話を進められる感じですね。)
ちなみにこれも前回修正しようと思って結局諦めていたんですが、「デオキシ」と「ジデオキシ」がややこしすぎるので、デオキシヌクレオチドをdNTP(dA, dC, dG, dTの4種類を総称してそう書きます)、ジデオキシヌクレオチドをddNTP(ddA, ddC, ddG, ddTですね)と書くようにします。明らかにその方がパッと見の可読性が高いので。
ということでDNA塩基配列解析=シーケンシング反応の説明ですが、前回はDNAを矢印で表していましたが、それはどう考えても分かり辛いので、今回は具体的な文字列を並べた形で見てみようと思います。その方が絶対に分かりやすいので。
その配列ですが、そもそもこの余談は「世界一甘いタンパク質・ソーマチンを合成しよう!」というネタから派生したものでしたし、せっかくなので既にクローニングの説明で何度か具体的な配列を見ていたこともあった、ソーマチン遺伝子のDNAを使って説明すると致しましょう。
まぁ実は最初っからこうしようと思ってたんですけどね(「矢印の概略図の次は、配列込みの図で具体的に見てみましょう」という流れで)、さっきも書いた通り、前回は時間がなくてできませんでした。
また、DNA合成酵素うんぬんの話が、あまりにも説明不足であったので、こちらももうちょい丁寧に説明していきたい限りですね。
では参りましょう。
こちらが、NdeIという制限酵素部位を使って挿入したソーマチン遺伝子の最初の十数塩基と、その上流の導入したプラスミドDNAの一部になります(遺伝子の最後はBamHIという制限酵素サイトを使っていますが、下流すぎてこの図では見えないので、まぁここではどうでもいいでしょう)。

やっぱり、無駄にアルファベットが並んだ図も一瞬「うっ…」と思うかもしれませんが、詳しくはこの辺の記事で見ていたので、意味不明な場合はそちらをご覧になっていただけると助かります。
まずDNAの配列を読む目的ですけど、pET-15bというプラスミドベクターに、NdeIサイト(とBamHIサイト)を使ってソーマチン遺伝子を挿入していたわけですが、以前も書いていた通り、「本当にソーマチン遺伝子が1塩基のミスもなく正しくプラスミドに入っているのか?」は、配列を確かめるまで何の保証もないわけです。
(いざ実験を最後まで進めて、なぜかソーマチンタンパク質が合成されない!→よく調べたら、業者に注文したDNAにミスが入っていた!時間と試薬を無駄にした、謝罪と賠償を請求する!!…とならないためにも、あらかじめ100%正しい確証を得ていくのは大切だということです。)
だから、(ソーマチン遺伝子の配列はもうデータベース上で知られているため)「知ってる遺伝子の配列を読むって、そんなことに何の意味が…?」と思われるかもしれないんですけど、ちゃんとこの目で「手持ちのプラスミドに、正しい配列が入っているか?」を確認する必要があるわけですね。
ここで一部だけ表示しているDNAは合計6000塩基以上のリング状のDNAだったのですが、前回書いた通り、DNAは高温に置くと、二本鎖状態だったのが手を離して、一本鎖ずつに分かれます。

鎖自身の結合(つながってる塩基間)は非常に強いので、それがバラバラになることは決してないけれど、鎖同士(二本の鎖間)の結びつきは弱いものなので、高温下に置くことで一本ずつに分離する…というのは前回書いた通りです。
ここで、最初のトリッキーなポイント、「配列を読みたい領域の上流に位置する、十数塩基の短いDNA」こと、プライマーを同じチューブに混ぜておく(ステップ2としていますが、実際は最初っからこの一本鎖DNAをチューブに混ぜてある感じですが)という話の登場ですね。

このプライマーは、自分の読みたい領域に応じて、あらかじめ自分で購入しておくものになります。
つまり、「読みたい領域の少し上流で、相棒の鎖(図でいう青い鎖)と結合する(A⇔T、C⇔Gが手をつないで二本鎖を形成する、いわゆる弱い結合ですが)十数塩基のDNA」ですが、その部分の配列を元に、自分で注文するわけです(これも、ソーマチン遺伝子購入に使った、DNA合成会社が安く販売しています)。
(この例なら、青い鎖の5'-ATGGCTGCCGCGCGG-3'の部分に結合する、5'-CCGCGCGGCAGCCAT-3'という15塩基のDNAですね。)
ということで、今からDNAの配列を読みたいのに、実は、その上流の配列をあらかじめ知っていなければいけないという謎な状況であるといえるんですね。
既知の配列のプライマーを使って、既知の配列の遺伝子をシークエンシングするという、初めて聞いたときは正直「お前は一体何やっとん?」と思う話になってるかもしれないんですけど、結局、サンガー法というのは「全く未知のDNAの配列を知る」のではなく、主に「どんなものか大体分かってるDNAの、実際の配列をチェックする・検証する」という意図で用いられるものだ、というと話が通じるでしょうか。
(先ほど書いた通り、プラスミドに挿入したソーマチン遺伝子が、1塩基のミスもなく正しい配列かどうかを確認する、的な。)
僕も初めて習ったときは、ウェスタンブロッティングの実験意図が不明だったのと同様、「は?今からDNAを調べるのに、知ってる配列が必要?知ってるなら、調べる必要ないじゃん!」とか思ったんですけど、結局そういう仕組みの実験になっている(未知のものを知るというより、いわば単なる確認実験)、ということを把握するのが理解への近道かもしれませんね。
もうちょい補足が必要な気もするのですが、いずれまた触れるとして、とりあえず話を進めましょう。
この、「プライマーが結合して、部分的に二本鎖になったDNA」が用意できたら、満を持してDNA合成酵素の出番なわけです。
前回はこれについての説明もかなり言葉足らずだったかもしれませんが、「DNA合成酵素」といってもDNAをゼロから合成するのではなく、チューブの中にあるヌクレオチドを使って、プライマーに連結させていくことでDNAを伸長する(ある意味「合成する」ともいえる)役割をもっているのが、この酵素なんですね。
(つまり、この酵素はDNAのコピーを作れるということですけど、そのコピー鎖を作るためには、反応の取っ掛かりとして、プライマーが結合した「一部が二本鎖になっている」DNAが必要になる、といえる感じですね。)
絵を見れば、きっと仕組みがご理解いただけると思います。

とても分かりやすい!(自画自賛)
結局、DNA合成酵素というのは、1塩基ずつデオキシヌクレオチド(dNTP)を取り込んで、プライマーを1塩基ずつ伸ばしていく酵素だということですね。
最終的には、こうなるわけです。

ここで表示されている範囲よりももっと先まで、延々とDNAを伸ばし続けてくれるのが、DNA合成酵素なのです。
なお、分かりやすく色を分けましたが(参照する相棒の鎖が青、プライマーが緑、新しく取り込まれた塩基が赤)、実際は、どれも同じデオキシヌクレオチドがつながっただけの、同じDNAの鎖ですね。
(あとちなみに、取り込まれた後、鎖の中ではdAとかdTではなくAとかTと表示していますが、これは単に見やすさのためにdを省略しているだけです。)
図の最後に書いた通り、最近の優れた酵素は、1秒で1000塩基も伸ばすことができるぐらい、凄まじい速さのものも売られています。
…と、仕組みというか流れについてはこの図でご理解いただけると思うんですけど、疑問に思うとしたら、「それを可能にしているメカニズム」といいますか、「は?何でそんなことが可能なん?酵素には目がついてるの?塩基を取り込んでつなげるって、こいつには手か何かがついとんのか?」みたいな点かと思うですけど、まぁこれは何度か書いている通り、にわかには信じがたいけれど、実際にそれを可能にしているのがタンパク質という有能分子だということですね。
もちろん、タンパク質には目も手も脳もないので、誰が操っているというわけでもありません。
ランダムに動き回っている分子同士がたまたま接触し、そこで捕まえた分子に対して酵素特有の機能を発揮できるという、いわば最初の接触は偶然に依存したものでしかない形なため、実際酵素を使ってる立場からしても、正直改めて考えると「本当にそんなこと可能か?」と思えなくもないんですけど、マジで、チューブの中にDNAとプライマーとdNTPと合成酵素とを混ぜたら、本当に完璧にDNA合成が進行するので、「信じられないかもしれないけど、もう何千回とやっているが、どの酵素でも確実に期待通りの反応がつつがなく実行されている。分子の力を信じろ」としかいえない感じですね。
(なお、その「分子の運動」は熱が重要なので、反応の温度は実際極めて重要なファクターになっています。混ぜた後に冷凍庫に置いたりしたら、反応は全く進行しません!)
さて、DNA合成酵素はともかく、シークエンシングでのポイントは、ddNTPによる反応の停止ということでした。
改めて、ddNTPが反応液中に存在すると、こうなるわけですね。

DNA合成酵素は、dNTPとddNTPの区別を付けられないので、ランダムにどちらかを取り込んでしまいます。
この性質を利用して、(前回もちょろっと書いていましたが)dNTPとddNTPとを絶妙な割合で混ぜることで、「一部のDNAはdNTPを取り込んで伸長が進むが、一部のDNAはddNTPを取り込んで伸長がストップする」という状況を生み出すことができるわけですね。
結果、こうなります。

「そんな上手いこと全部の場所で止まるの?たまたまddNTPが全く取り込まれないやつがいたら、どーする?!」と思われるかもしれませんが、プライマーは何百万何千万分子と加えるので、多少の量の違いはあれど、ほぼ確実にあらゆる塩基でストップした断片ができあがります。
…と、ここまでが前回も書いていた話でした。
「長さと、末端の塩基とを対応させることができれば、配列の解読(シークエンシング)ができる!!」ということで、それをどうやって対応させるか、という所で次回へ続く…って感じだった形ですね。
既に前回と同じぐらいの長さになっている気もしますが、まぁ案外簡単な話でしかないので、検出の方法について進めちゃいましょう。
サンガーさんが考えた方法は、「4つのチューブに、それぞれ1種類のddNTPしか入れずに反応を進めてやろう。この反応産物を、ゲルを用いて電気泳動で大きさごとに分離すれば、配列が読めるはずだ!」というものでした。
図を見れば恐らくサンガーさんの考えが一発でご理解いただけることでしょう。

つまり、ddATPを入れて反応をさせたチューブでは、Aの場所でしかDNA伸長が止まらないので、ここで止まっているDNAの末端がAであることは確定しているわけですね。
これを下(泳動の先端)から順番に読めば、バッチリ、DNAの配列解読完了なわけです。
流石はノーベル賞2度受賞のサンガーさん、賢い!
…しかし、これは1つのDNAを読むのに4つの反応が必要で、それに応じて流すゲルも大型のものが必要になるので、若干不便なのです。
そこで、後年、技術の発展とともに、さらに賢く便利で優れものな、素晴らしい技法が生まれました。
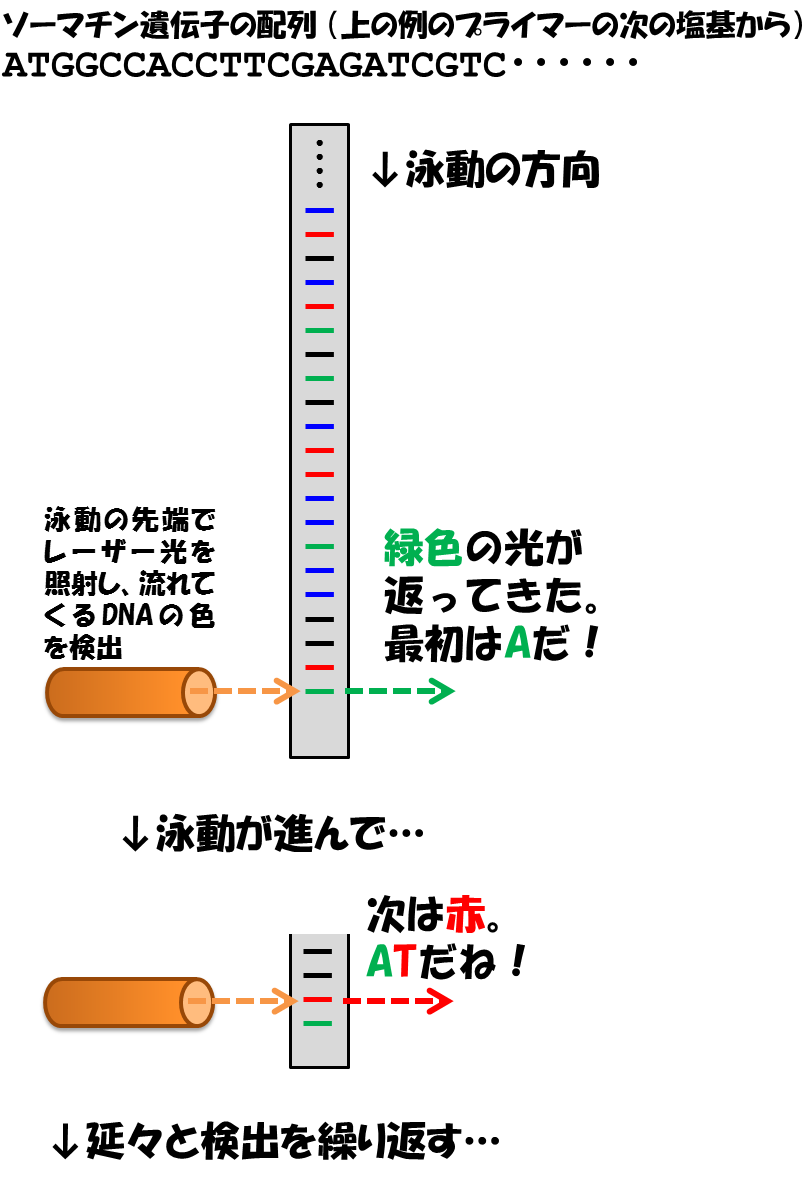
それが、今現在でも最もよく使われている方法、蛍光色素をddNTPにつける方法になります!

色素=dye、ddNTPは反応を終結させるのでterminatorと呼ばれ、この方法はダイターミネーター法と呼ばれていますが、まぁサンガー法の亜流なので、普通にSanger sequencingと呼ばれることの方が多いですけどね。
画像を見ればもう説明不要でしょう、4種のddNTPに、それぞれ異なる色の蛍光色素をつなげて、どのddNTPが取り込まれたかが色で分かる仕組みになっているわけです。
これを、普通のゲルではなく、キャピラリーという(まぁ日本語で「毛細管」という意味の語ですが)特殊な微小装置の中にDNAを流すことで、レーザー光を用いた高解像度での分離分析が可能となっています。
これも、図を見ていただくと、もう一発でしょう。
現在DNAシークエンシングサービスを受託している企業は山のようにありますが、ほぼ全ての企業が、一番簡単な日常用途のシーケンシングとして、このダイターミネーター法を用いています。
実際、僕自身今でも日常的に使っていますし、せっかくなのでちょうどつい先日、実際にDNAを業者に送って返ってきた結果をご紹介してみましょう。
色々反応の原理などまとめましたが、実際には、手元にある「配列を読みたいDNA」に、「自分の読みたい領域の上流と結合するプライマー」を混ぜて、シーケンシングサービスを提供している会社にサンプルを送るだけなので、仕組みは知らなくともDNAとプライマーを送るだけで結果が返ってくるという便利な世の中になっています。
業者の方でも、ほぼ全てが自動化されて、受け取ったDNAを機械にかけるだけだと思うので、まさに多くの方がイメージしていらっしゃるであろう、「DNAを機械にかけたら、自動的にカタカタカタッと文字列が表示される」というのと、ほぼ遜色ないレベルの便利さといえますね。
具体的には、pcDNA3.1(+)という、大腸菌ではなくヒト細胞に導入する用途で汎用されているプラスミドベクターがあるのですが、その制限酵素部位に遺伝子DNAを入れて、配列をチェックした結果になります。
こんな感じ…

業者から送られてきたシーケンシングファイルをSnapGeneというこないだもちょろっと紹介していた遺伝子解析ソフトウェアで開いた形ですが、GGGCCCがマルチクローニングサイト(制限酵素が多数あって、自分の好きな遺伝子を入れられる便利な部位)の最後の部分=ApaIサイトで、そこから下流はpcDNA3.1(+)の配列だということですね。
(つまり、この上流が、自分の入れた遺伝子の配列ですが、そこを見せるとやってる研究が分かってしまうので、プライバシー保護(?)で伏せている感じです。)
こんな感じで、美しいピークとして、4色の色で配列が分かります。
ちなみに、こちらがpcDNA3.1(+)のデータベースの配列(プラスミドマップ)ですが…

先ほど貼った、実際にシーケンシングして読んだ配列と見比べてみると、まさに、1塩基の違いもなく、完全にデータ通りの配列が読めていることがお分かりいただけるように思います。
ちなみに、シーケンスファイルのピークレポート図で、362番目のGがたまたまハイライトされていましたが、Quality: 61となっていましたね。
これは、Phredのクオリティ・スコアと呼ばれるもので…

スコアが60あれば99.9999%正しい、つまりほぼ間違いないものと断言できるものなわけですね。
(まぁ、あのGが間違ってる可能性は、スコアなど見ずとも、見た目的に絶対にないと断言できますが。)
全体的に大変美しいピークで、これは、自分の用意したプラスミドのクオリティがとても良かったことの裏返しといえましょう(自画自賛)。
実際、下手な人がやると、ノイズまじりのピークが得られて(スコアも低い)、せっかく読んだのに所々微妙で正しい配列が分からない…なんてこともあり得る感じです。
なお、ピークレポート右上に1127 basesと表示されていた通り、一応1127塩基読めた形ですが、最後の方はもうぐっちゃぐちゃで、信頼できる配列にはなっていません。
これがピークレポートファイルの終わりの部分…

Quality: 8とか笑えますね(実際、これホンマCか?と思えます)。
ただ、僕の読みたかったのは自分の入れた遺伝子=最初の300塩基の部分だけなので、もうこの辺は一切不要な情報なので問題ありません(ただどうでもいいプラスミドベクターの配列が表示されてるだけ)。
一般的にサンガー法で読めるのは1000塩基程度が限界といわれていますが(ちょうど、そのぐらいでddNTPが枯渇するのか、キャピラリー分析の限界なのか、シグナルがでたらめになってくる感じですね)、これも、汚いDNAだと500塩基ぐらいでもうしっちゃかめっちゃかなピークになるので、改めて、自分の用意したDNAは1000塩基もキレイに読めてて、素晴らしいクオリティのものだったといえる感じですね。
ちなみに、ほとんどの研究室はDNAの配列解析を外部の業者などに委託していると思うんですけど、僕が大学生時代にいた研究室はかなりリッチで、自前でシークエンシング機械をもっていました。
なので、自分で色素を混ぜてキャピラリー電気泳動して……というのも経験したことがありますけど、当時は学生だったしあんまりコストみたいなのを意識していなかったので、全く同じDNAを「念のため…」とかいって8個とか配列解析していたんですけど、何気に、試薬代とかのランニングコストもバカ高いのがシークエンシング反応なのです。
今委託しているのは大体1サンプル4ドルぐらいですけど、多くの業者では500円600円、下手したら1000円ぐらい取る所もあるので、無知な学生だったとはいえ、当時はクッソ無駄使いしてしまっていたなぁ…と冷や汗ものというか申し訳なさでいっぱいです。
(まぁ別にそれでも何もいわれなかったぐらい予算の潤沢にある研究室でしたし、幸いそんなにシーケンシングするタイプの学生でもなく、滅多にやらなかったい実験だったので、ヤバイレベルの無駄使いではなかったんですけどね。)
…ってな所で、サンガー法によるDNAシークエンシングの仕組みについて、実例も交えて紹介してみましたが、このぐらいで紹介しておきたかった内容は出せたように思います。
(あぁ、プライマーについて、書き足りないところもあったかもしれませんが、いずれ補足したいですね。)
恐らく、以上の話を踏まえて、「DNAの配列を読むためにはプライマーが必要らしいけど、じゃあ、全く未知のDNAを読むことはできないわけ?」という疑問点が浮かぶ方もいらっしゃるように思うのですが、実際、それは余裕で可能です。
特に、次世代シーケンシングと呼ばれる技術で、大量のDNAを、未知の領域だろうとなんだろうと有無をいわさず大量に読み込む技法が確立&発展し続けており、現在盛んに使われています。
次回は、その次世代シーケンス技術を……と思いましたが、次世代だけあってあまりにもややこしく複雑なので、流石に仕組みに深入りするのはやめようと思いますが、めっちゃ簡単に、次世代シーケンシングでできることぐらいをまとめてみようかなと思っています。